- Blog
Global implications of the implementation of IDMP

Identification of Medicinal Products (IDMP) refers to a suite of International Organization of Standardization (ISO) standards whose objective is to define, characterize, and uniquely describe regulated medicinal products. This definition will support all regulatory documentation and processes concerned with medicinal products throughout their life-cycle, from development through to authorization, post-marketing management, and final withdrawal from the market. The suite is led by ISO 11615, the Medicinal Product and Packaged Product standard, which provides the pattern to uniquely describe each product and each package as it is authorized. The other standards support by contributing concepts: Substance Information (ISO 11238), Dose Form or Packaging Concepts (ISO 11239), Units of Measurement (ISO 11240), and Pharmaceutical Product information (ISO 11616).
Although now delivered through ISO, IDMP originated in the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use initially for pharmacovigilance. Much of the stimulus for implementation has occurred in Europe, led by the European Medicines Agency (EMA), motivated by the regulations for Eudravigilance, which include implementing regulation 520/2012 for medicinal product terminology that specifically states the use of the ISO IDMP standards, and prior to their implementation, the xEVMPD (Article 57) legislation.
However, having been through the ISO process, the IDMP standards are international standards, and to truly fulfill their aim of supporting all regulatory documentation and processes concerned with medicinal products throughout their life-cycle, their implementation must be global. So what do we see if we look more widely?
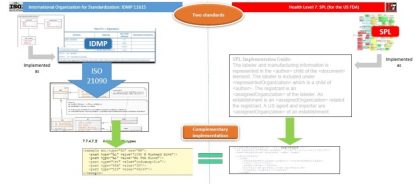
In many respects, the US Food and Drug Administration (FDA) has already commenced implementation of IDMP, through its Structured Product Label (SPL) specification as part of its Guidance for Industry on Electronic Submissions (in PDUFA – the Prescription Drug User Fee Act). Looking at the content of an SPL submission, a significant proportion (more than 80%) of the data items required by the 11615 standard are already present within the SPL specification, with the main difference being in how those data items are described. For example, indications may be given in text in SPL, whereas they must be coded in IDMP; and for the items that are coded, the controlled terminology that is currently in use differs. However, with a commitment to move towards using the Global Ingredient Archival System (GINAS) and its G-SRS software, which maintains and makes available global coded concepts for ingredient substances (thereby implementing ISO 11238), and the EDQM controlled terminology for dose forms (units of presentation, e.g., the implementation of ISO 11239 within the SPL, etc.) that mismatch diminishes significantly.
Bearing in mind that the first submission-based implementation of IDMP in Europe will use the SPL specification, as described in the in-development ISO DTS 20443 Technical Specification, the correspondence between what is already done in the US and what is being implemented in Europe becomes even greater — which, of course, is how it should be for a global standard to facilitate information-sharing.
Figure 1: Diagram showing the parallels for implementation of ISO IDMP and SPL
Many other regulatory agencies around the globe are actively looking at when and how to implement the IDMP standards within, or in addition to, their existing processes, usually in a stepwise fashion, so that the global objectives of IDMP can be achieved. The International Pharmaceutical Regulators Forum (IPRF) Management Committee has explicitly supported engagement in IDMP, with agreement in principle for a work group to be formed to take this forward.
The international implementation has significant value for pharmaceutical companies themselves. The fundamental objective of the IDMP standards is to facilitate meaningful sharing of information. Having data defined using the same patterns and using the same controlled terminology in all areas of the life-cycle for all medicinal products means that processes can be harmonized, KPIs can be more appropriately applied, and headquarters and affiliates can far more easily communicate and share with one another.
Global for the enterprise
Just as implementation of IDMP will eventually affect every regulatory agency in the world, so implementation of IDMP will gradually ripple out and affect every part of the enterprise of a pharmaceutical company. Despite IDMP having originated in pharmacovigilance — propelled by the need to be able to accurately and uniquely identify suspect products and concomitant medications globally to find “the needle in the haystack"1 for adverse reactions — most companies are looking to implement, at least initially, through Regulatory Affairs, often employing a master data management strategy. But once implemented, the critical master data should be available to all areas of the enterprise as well as RA and PV.
Here are just a couple of examples:
- Clinical will be involved, providing information about investigational products, but also having access to the this global medicinal product terminology, which, for example, means that the concomitant medications of study subjects can be accurately and globally described; inclusion and exclusion (I/E) criteria that involve medications (just under 20% of all I/E criteria) can be accurately described – and indeed even coded – so that investigators can have systems support in the selection of subjects for studies, focusing and standardizing recruitment across sites.
- Supplies and logistics will be involved, providing information about the batches of products released in different jurisdictions, supporting anti-counterfeiting measures (including through links with GS1), and helping to avoid critical product shortages. Should a batch-recall be necessary for any reason, the increased accuracy of description and supporting information available from IDMP should help manage that critical process more effectively for all.
Can implementation of IDMP standards be any wider than this?
Although the stated objective of the IDMP suite of standards is to support all regulatory documentation and processes concerned with medicinal products throughout their life-cycle, there is another area where IDMP implementation can and is having an effect – in patient care itself, positively contributing to the “bench-to-bedside" vision of clinical research and translational medicine. Various initiatives are looking at how to use IDMP medicinal product data directly in patient care:
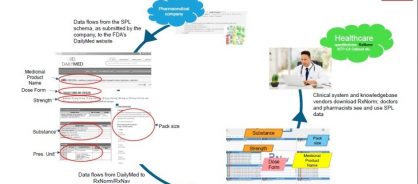
- In the US, SPL data from authorization holders is sent directly on from the FDA to the National Library of Medicine (NLM) DailyMed website, where it is displayed, using a “style sheet" overlaid onto the XML submitted by the company, and from there the same data is sent on through where it is one of the contributors to the RxNorm system. RxNorm is a code system for medicinal products that is used in physician systems and pharmacies to support interoperable healthcare in the US, particularly for “Meaningful Use" certification.
- In the European Union, IDMP product data is being actively considered to support the openMedicine initiative, initially for cross-border prescription support, but in the future for the Electronic Patient Summary initiative.
- In many other countries, through various national initiatives, closer working relationships are being forged between national healthcare terminology providers and regulatory agencies from whom the authorized data originates as a result of the IDMP standards.
Figure 2: Diagram showing how SPL information flows through to clinicians' systems
In conclusion, the implementation of the IDMP suite of standards will potentially have positive and far-reaching value, not only for pharmaceutical companies and regulatory agencies, but also for the global provision of healthcare.
1. Edwards, I. Adverse drug reactions: finding the needle in the haystack. BMJ 1997; 315:500
Share